


作者:i医法律服务团队 | 2022.06.22
药物临床试验是药品批准上市的关键环节,是评价药物有效性和安全性的关键步骤,真实、规范、完整的临床试验数据,是药品安全性和有效性的源头保障。随着监管力度的加强,实务中,药物临床试验各参与方,无论是申办方、合同研究组织或者临床试验机构、研究者,都需要不断强化临床试验数据合规管理的理念,提升对法律风险的防范意识。
康达律师事务所i医法律服务团队资深律师基于在医疗健康和生命科学领域的实务积累,对药物临床试验数据数据管理中广受关注的真实性和合规性问题进行综合性分析,以期为相关从业者提供参考。
2022年6月1日,国家药品监督管理局发布《2021年度药品审评报告》(以下简称“报告”),其中指出,2021年,国家药品监督管理局药品审评中心(以下简称药审中心)受理注册申请11658件,同比增长13.79%,同时公布了2017-2021年注册申请受理量,呈现逐年增长的态势,详见图1。
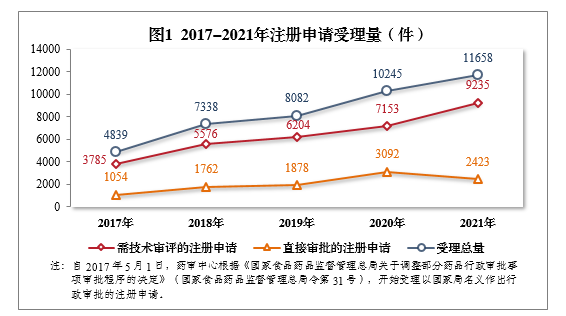
图1,2017-2021年药审中心注册申请受理量
(图片来源:国家药品监督管理局《2021年度药品审评报告》)
报告第四章《关于药品注册申请存在的主要问题及分析》中指出,2021年,药品注册申请经技术审评后审评结论为不批准/建议不批准的注册申请542件,其中,359件属于因申请人未能在规定时限内补充资料的情形,占全年不批准/建议不批准总量的66.3%,包括中药9件、化学药349件、生物制品1件;183件注册申请主要存在申报资料无法证明申请注册药品的安全性、有效性或质量可控性等缺陷问题,包括中药14件、化学药126件、生物制品43件。主要包括六大问题,分别为研发立题方面、有效性方面、安全性方面、质量可控性方面、合规性方面和其他方面。其中,合规性方面常见于经注册核查和注册检验的注册申请,具体包括注册核查中发现研究数据存在真实性问题、注册核查中发现其他影响产品质量的重大缺陷;注册核查抽样检验不合格。
报告中亦指出,总体上看,2021年注册申请存在的主要问题,在分类、具体表现等方面与往年具有较大的相似性,但也出现了一些变化,比如因合规性问题而未获批准的注册申请数量较往年有减少趋势。该趋势变化与临床试验管理增强,核查检验协调机制协调发展密切相关,更得益于2015年以来对临床试验数据自查核查工作的有序开展。
随着监管力度的加强,实务中,药物临床试验各参与方,无论是申办方、合同研究组织或者临床试验机构、研究者,都需要不断强化临床试验数据合规管理的理念,提升对法律风险的防范意识。在此,康达律师事务所i医法律服务团队基于在医疗健康行业多年的法律服务经验,结合相关法律法规,对药物临床试验中的数据真实性和合规性要点进行解读和分析,以期为相关从业人员提供参考。
药物临床试验是药品批准上市的关键环节,是评价药物有效性和安全性的关键步骤,真实、规范、完整的临床试验数据,是药品安全性和有效性的源头保障。
2015年以来,我国围绕着质量和创新两个核心主题推进药品审评审批制度改革,2019年新修订的《药品管理法》正式实施,2020年新版《药品注册管理办法》和《药物临床试验质量管理规范》(GCP)开始执行。随着一系列新修订和配套文件的颁布,新的监管政策落地,临床试验监管方式上发生较大的变化,药品审评审批工作对药物临床试验监管的要求正在逐步提升,药品监管部门不断加强药物临床试验数据核查力度。
2015年7月22日,为落实党中央、国务院“用最严谨的标准、最严格的监管、最严厉的处罚、最严肃的问责,确保广大人民群众饮食用药安全”的要求,国家食品药品监督管理总局发布《关于开展药物临床试验数据自查核查工作的公告》(2015年第117号),决定对待审药品注册申请开展药物临床试验数据核查。 其中指出,自查的内容包括:(一)核对锁定的数据库与原始数据一致性,统计分析以及总结报告数据与原始记录及数据库的一致性;数据锁定后是否有修改以及修改说明等。(二)生物样本分析测试仪器(如HPLC、LC-MS/MS)等主要的试验仪器设备运行和维护、数据管理软件稽查模块(Audit trail)的安装及其运行等。(三)各临床试验机构受试者筛选、入组和剔除情况,受试者入选和排除标准的符合情况,抽查核实受试者参加临床试验的情况。(四)临床试验方案违背例数、剔除例数、严重不良事件例数等关键数据;医院HIS系统等信息系统中的受试者就诊信息、用药及检查化验的临床过程情况等。(五)试验药物和对照药品的生产或购进、检验、运输、保存、返还与销毁以及相关票据、记录、留样等情况。(六)生物样本的采集过程及运送与交接和保存等记录;生物样本分析方法确证,生物样本分析过程相关的记录以及样本留样情况。(七)有关方在临床试验项目中主要职责的落实情况、合规情况。 后续为持续推进数据核查工作,国家食品药品监督管理总局和国家卫生计生委又陆续发布相关通知文件,指导数据自查核查工作的开展。例如,2015年11月13日国家卫生计生委发布《关于进一步做好药物临床试验机构自查工作的通知》,食品药品监管总局分别于2015年12月17日发布《关于进一步加强药物临床实验数据自查核查的通知》,于2016年3月28日发布《关于印发药物临床试验数据核查工作程序(暂行)的通知》,于2016年6月2日发布《关于药物临床试验数据自查核查撤回品种重新申报有关事宜的公告》(2016年第113号),于2017年5月25日发布《关于药物临床试验数据核查有关问题处理意见的公告》(2017年第63号)。此外,为了方便临床试验参与者了解数据核查工作,CFDI公布了临床试验数据核查流程,如下图2。
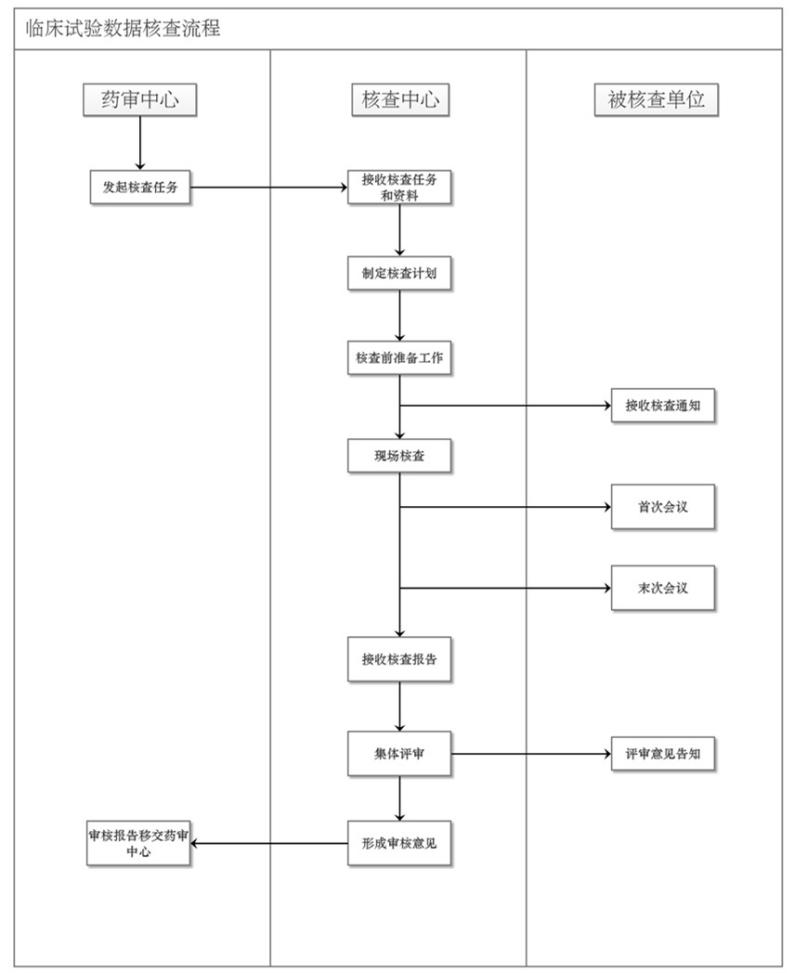
图2,临床试验数据核查流程
(图片来源:CFDI官网办事指南-现场检查工作流程)
2021年12月17日,按照《药品注册管理办法》规定,为明确药品注册核查实施的原则、程序、时限和要求,规范药品注册生产现场核查和上市前药品生产质量管理规范检查衔接工作,国家药品监督管理局食品药品审核查验中心组织制定并发布了《药品注册核查工作程序(试行)》《药品注册核查要点与判定原则(药理毒理学研究)(试行)》《药品注册核查要点与判定原则(药物临床试验)(试行)》及《药品注册核查要点与判定原则(药学研制和生产现场)(试行)》和《药品注册生产现场核查和上市前药品生产质量管理规范检查衔接工作程序(试行)》,自2022年1月1日起施行。 药物临床试验现场核查是药品注册研制现场核查的一种,国家药品监督管理局药品审评中心(CDE)负责药品技术审评,而现场核查通常由国家药品监督管理局食品药品审核查验中心(CFDI)组织实施。 《药品注册核查要点与判定原则(药物临床试验)(试行)》指出,药品注册现场核查(药物临床试验)的目的主要是通过对注册申报资料与临床试验的原始记录和文件的核对和/或实地确证,评价试验实施、数据记录和结果报告是否符合试验方案和药物临床试验相关法规,核实相关申报资料的真实性、一致性,同时关注受试者保护。 临床试验部分现场核查要点在于临床试验许可与条件、伦理审查、临床试验实施过程、试验用药品管理、生物样品管理、中心实验室及独立评估机构、临床试验数据采集与管理和委托研究等方面。生物样品分析部分现场核查要点在于生物样品分析条件与合规性、生物样品分析实验的实施、记录的管理等方面。
2017年发布的《关于药物临床试验数据核查有关问题处理意见的公告》中对药物临床试验数据核查中发现的有关问题的处理意见予以公告。公告中强调了药物临床试验参与者的三大主体责任。其中指出,申请人是药品注册的申请者和权利人,必须保证注册申请中临床试验数据的真实、完整和规范,监督临床试验项目的实施,对所报申请资料及相关试验数据可靠性承担法律责任。研究者受申请人委托具体实施临床试验项目,必须保证试验行为符合GCP规定,保证试验数据真实、完整、规范及可溯源,对临床试验数据真实性、完整性、规范性承担直接法律责任。临床试验机构是药物临床试验项目直接管理者,对临床试验数据的真实性、完整性和规范性负有管理监督责任。临床试验合同研究组织受申请人委托,承担临床试验相关工作,对临床试验数据真实性、完整性、规范性承担法律及合同约定的责任;对其出具的相关报告和数据承担直接法律责任。 对于数据造假的认定予以明确,具体包括以下情形:(一)编造或者无合理解释地修改受试者信息以及试验数据、试验记录、试验药物信息;(二)以参比制剂替代试验制剂、以试验制剂替代参比制剂或者以市场购买药品替代自行研制的试验用药品,以及以其他方式使用虚假试验用药品;(三)隐瞒试验数据,无合理解释地弃用试验数据,以其他方式违反试验方案选择性使用试验数据;(四)瞒报与临床试验用药相关的严重不良事件,瞒报可能与临床试验用药相关的严重不良反应事件;(五)瞒报试验方案禁用的合并药物;(六)故意损毁、隐匿临床试验数据或者数据存储介质;(七)其他故意破坏药物临床试验数据真实性的情形。 而对于上述不同主体的数据造假行为,国家食品药品监督管理总局明确了处理原则,列表如下:
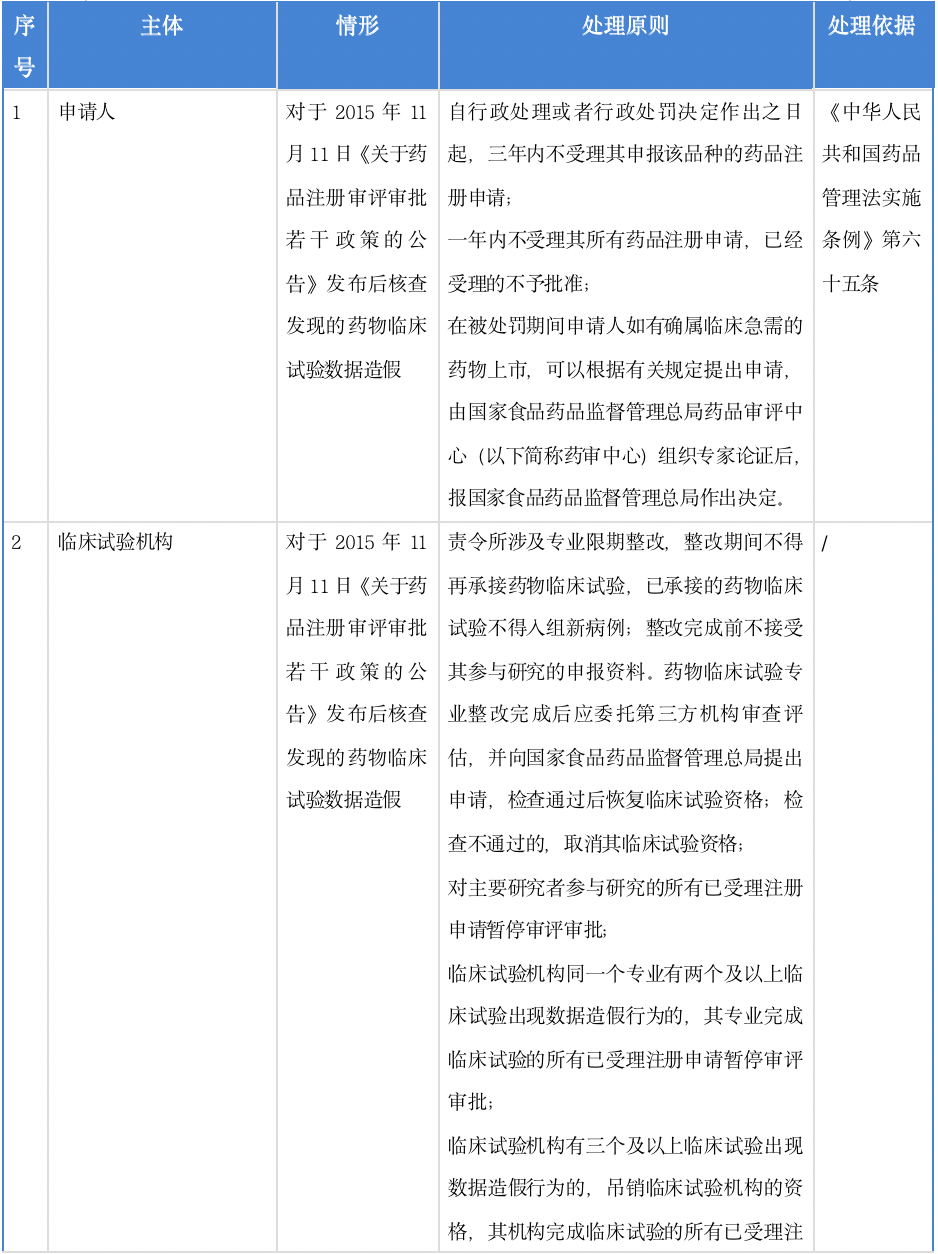

我国2019年修订的《药品管理法实施条例》第六十五条与2016年修订的《药品管理法实施条例》保持一致,其中指出,药品申报者在申报临床试验时,报送虚假研制方法、质量标准、药理及毒理试验结果等有关资料和样品的,国务院药品监督管理部门对该申报药品的临床试验不予批准,对药品申报者给予警告;情节严重的,3年内不受理该药品申报者申报该品种的临床试验申请。
除了上表中的法律规定,2020年7月1日起施行的《药品注册管理办法》第一百一十一条规定,在药品注册过程中,提供虚假的证明、数据、资料、样品或者采取其他手段骗取临床试验许可或者药品注册等许可的,按照《药品管理法》第一百二十三条处理。2019年修正的《药品管理法》第一百二十三条指出,提供虚假的证明、数据、资料、样品或者采取其他手段骗取临床试验许可、药品生产许可、药品经营许可、医疗机构制剂许可或者药品注册等许可的,撤销相关许可,十年内不受理其相应申请,并处五十万元以上五百万元以下的罚款;情节严重的,对法定代表人、主要负责人、直接负责的主管人员和其他责任人员,处二万元以上二十万元以下的罚款,十年内禁止从事药品生产经营活动,并可以由公安机关处五日以上十五日以下的拘留。
对于临床试验数据不完整、不规范,但是暂未构成数据造假的情形,国家食品药品监督管理总局在《关于药物临床试验数据核查有关问题处理意见的公告》亦明确了处理原则:对临床试验数据不完整、不规范,不足以证明药品安全性和有效性的,其注册申请不予批准;仅存在数据不规范,通过补充资料可以完善的,由国家食品药品监督管理总局要求申请人一次性补充,补充后按程序进行审评审批。
另外,为了推动临床试验数据自查,国家食品药品监督管理总局在《关于药物临床试验数据核查有关问题处理意见的公告》指出,主动开展自查,主动报告问题,主动撤回申请的,可以免除行政处罚。就申请人而言,在核查前主动撤回注册申请的,可以免于行政处罚;申请人可以按照《关于药物临床试验数据自查核查撤回品种重新申报有关事宜的公告》(国家食品药品监督管理总局公告2016年第113号)有关要求重新开展或者补充完善临床试验。临床试验机构、合同研究组织自查发现数据不真实的,主动将具体品种、申请人的名称以及不真实的具体问题向国家食品药品监督管理总局和所在地省级食品药品监督管理局报告,同时告知申请人的,可以免除行政处罚。
除了药物临床试验数据核查自查的相关规定外,2022年开始施行的《药品注册核查要点与判定原则(药物临床试验)(试行)》,对研究过程中原始记录和数据进行核实、实地确认,亦规定了核查结果判定原则:
(1)经核查确认发现以下情形之一的,核查认定为“不通过”:
1. 编造或者无合理解释地修改受试者信息以及试验数据、试验记录、试验药物信息;
2. 以参比制剂替代试验制剂、以试验制剂替代参比制剂或者以市场购买药品替代自行研制的试验用药品,以及以其他方式使用虚假试验用药品;
3. 隐瞒试验数据,无合理解释地弃用试验数据,以其他方式违反试验方案选择性使用试验数据;
4. 瞒报可疑且非预期严重不良反应;
5. 瞒报试验方案禁用的合并药物;
6. 故意损毁、隐匿临床试验数据或者数据存储介质;
7. 关键研究活动、数据无法溯源;
8. 申报资料与原始记录不一致且影响结果评价;
9. 其他严重数据可靠性问题;
10. 拒绝、不配合核查,导致无法继续进行现场核查;
11. 法律法规规定的其他不应当通过的情形。
(2)对研究过程中原始记录和数据进行核实、实地确认,未发现问题或发现的问题不构成以上不通过情形的,核查认定为“通过”。其中发现的问题对数据质量和可靠性可能有影响的,需审评重点关注。
对于核查认定为“不通过”的,依据《药品注册管理办法》第九十二条规定,不予批准药品注册申请。《药品注册管理办法》第九十二条指出,药品注册申请有下列情形之一的,不予批准:……(六)药品注册过程中认为申报资料不真实,申请人不能证明其真实性的;(七)药品注册现场核查或者样品检验结果不符合规定的。
《药品注册核查要点与判定原则(药物临床试验)(试行)》中“不通过”的情形中部分与数据造假行为的认定一致,药物临床实验数据专项核查工作也逐步践行至常规药品注册核查过程中。在注册核查过程中出现的不合规情形若上升到数据造假层面的,依然应按照相关法律法规予以处罚。
对于药物临床试验参与方的刑事责任问题,为依法惩治药品、医疗器械注册申请材料造假的犯罪行为,维护人民群众生命健康权益,2017年最高人民法院、最高人民检察院曾发布了《关于办理药品、医疗器械注册申请材料造假刑事案件适用法律若干问题的解释》(以下简称《解释(2017)》),对临床试验中的不同犯罪行为及刑事责任予以明确。其中指出,在药品注册申请中,故意提供、使用虚假的临床试验报告及相关材料的,依据行为主体与具体情节,临床试验参与者可能涉及故意提供虚假证明文件罪,生产、销售假药罪,受贿罪,非国家工作人员受贿罪等等。 随着2022年3月6日最高人民法院、最高人民检察院发布的《关于办理危害药品安全刑事案件适用法律若干问题的解释(2022)》(以下简称《解释(2022)》)的施行,2017年发布的上述解释随之废除。《解释(2022)》对药品相关的临床试验数据造假等相关行为的刑事责任进行了明确,主要依据刑法第一百四十二条规定的妨害药品管理罪予以处罚。 2021年3月1日,我国《刑法》(2020修正)开始施行,其中在第一百四十二条新增妨害药品管理罪,该条规定,违反药品管理法规,有下列情形之一,足以严重危害人体健康的,处三年以下有期徒刑或者拘役,并处或者单处罚金;对人体健康造成严重危害或者有其他严重情节的,处三年以上七年以下有期徒刑,并处罚金:……(三)药品申请注册中提供虚假的证明、数据、资料、样品或者采取其他欺骗手段的。 对于《刑法》第一百四十二条中具体情形,如“足以严重危害人体健康的”、“对人体健康造成严重危害”以及“或者有其他严重情节的”的认定,《解释(2022)》进行了进一步的解释。 《解释(2022)》第七条指出,实施妨害药品管理的行为,具有下列情形之一的,应当认定为刑法第一百四十二条之一规定的“足以严重危害人体健康”:……(六)在药物非临床研究或者药物临床试验过程中故意使用虚假试验用药品,或者瞒报与药物临床试验用药品相关的严重不良事件的;(七)故意损毁原始药物非临床研究数据或者药物临床试验数据,或者编造受试动物信息、受试者信息、主要试验过程记录、研究数据、检测数据等药物非临床研究数据或者药物临床试验数据,影响药品的安全性、有效性和质量可控性的。 《解释(2022)》第八条指出,实施妨害药品管理的行为,具有本解释第二条规定情形之一的,应当认定为刑法第一百四十二条之一规定的“对人体健康造成严重危害”。《解释(2022)》第二条指出,生产、销售、提供假药,具有下列情形之一的,应当认定为刑法第一百四十一条规定的“对人体健康造成严重危害”: (一)造成轻伤或者重伤的; (二)造成轻度残疾或者中度残疾的; (三)造成器官组织损伤导致一般功能障碍或者严重功能障碍的; (四)其他对人体健康造成严重危害的情形。 《解释(2022)》第八条指出,实施妨害药品管理的行为,足以严重危害人体健康,并具有下列情形之一的,应当认定为刑法第一百四十二条之一规定的“有其他严重情节”:……(三)药品申请注册中提供虚假的证明、数据、资料、样品或者采取其他欺骗手段,造成严重后果的。 此外,对于参与临床试验造假活动的不同主体的犯罪行为认定,《解释(2022)》第九条指出,明知他人实施危害药品安全犯罪,而有下列情形之一的,以共同犯罪论处:……(四)提供虚假药物非临床研究报告、药物临床试验报告及相关材料的。
随着“健康中国2030”规划纲要的稳步落实,为保障医疗行业的健康发展,我国对药品全生命周期的监管不断加强,临床试验作为评价药物有效性和安全性的关键步骤,相关从业者应强化全流程合规管理的理念,保障临床试验数据的真实性和准确性,积极防范法律风险,做到合法合规注册,真正造福患者。
地 址: 北京市朝阳区建外大街丁12号英皇集团中心8层,邮编:100022
电 话:(010) 5086 7666
传 真:(010) 5691 6450
地 址: 西安市雁塔区太白南路139号荣禾云图中心7层、15层,邮编:710061
电 话:(029)8836 0129
传 真:
地 址: 深圳市福田区中心四路一号嘉里建设广场1期19楼,邮编:518046
电 话:(0755)8860 0388
传 真:
地 址: 海口市美兰区碧海大道86号华彩·海口湾广场A座1008、1009,邮编:570208
电 话:(0898)6625 4181
传 真:(0898)6625 5316
地 址: 上海市黄浦区龙华东路325号博荟广场A座1206室,邮编:200023
电 话:(021)6390 1100
传 真:(021)6390 1010
地 址: 广州市天河区珠江东路32号利通广场29层2901室,邮编:510510
电 话:(020)3739 2666
传 真:
地 址: 杭州市上城区西子国际中心2号楼1501-1503室,邮编:310002
电 话:(0571)8577 9929
传 真:
地 址: 沈阳市和平区南湖街道青年大街390号皇朝万鑫大厦C座21层,邮编:110004
电 话:(024)2250 3388
传 真:
地 址: 南京市建邺区应天大街888号金鹰世界A座26层,邮编:210008
电 话:(025)8411 1616
传 真:
地 址: 天津市河北区海河东路78号茂业大厦2601室,邮编:300141
电 话:(022)2445 9827
传 真:
地 址: 菏泽市开发区人民路菏建·数码大厦B座西单元19层,邮编:274005
电 话:(0530)5566 148
传 真:
地 址: 成都市锦江区东御街18号百扬大厦1栋11层1101室,邮编:610020
电 话:(028)8774 7485
传 真:
地 址: 苏州市工业园区九章路69号理想创新大厦A幢12层,邮编:215316
电 话:(0512)6758 6952
传 真:(0512)6758 6972
地 址: 呼和浩特市金桥开发区昭乌达路宇泰商务广场A座11层1101室,邮编:010041
电 话:(0471)5166 277
传 真:
地 址: 九龍渡船街38號建邦商業大廈1樓5號室
电 话:(00852)2333 9989
传 真:(00852)2333 9186
地 址: 武汉市江岸区中山大道1627号中信泰富大厦20层,邮编:528451
电 话:(027)8261 8977
传 真:
地 址: 郑州市金水区郑东新区农业南路51号楷林中心10座12层,邮编:450046
电 话:(0371)8895 9887
传 真:
地 址: 长沙市雨花区芙蓉中路三段567号第六都兴业IEC32层,邮编:410021
电 话:(0731)8218 3551
传 真:(0731)8218 3551
地 址: 厦门市湖里区高林中路469号新景地大厦23层,邮编:361016
电 话:(0592)5211 009
传 真:
地 址: 重庆市江北区桂花街支路10号成大锦嘉国际大厦10层,邮编:400020
电 话:(023)6775 9966
传 真:
地 址: 合肥市蜀山区政务区华润大厦西塔B座30层,邮编:230071
电 话:(0551)62930997
传 真:
地 址: 宁波市鄞州区三眼桥街51号宁铸中心5号楼27层(宁波塔-27F),邮编:315199
电 话:(0574)8737 8737
传 真:
地 址: 济南市高新区舜泰北路舜泰广场933号博晶大厦25层2513室,邮编:250101
电 话:(0531)8828 5613
传 真:
地 址: 昆明市西山区融城优郡B幢10楼,邮编:650034
电 话:(0871)6517 9639
传 真:
地 址: 南昌市红谷滩区红谷中大道1391号华皓中心53层,邮编:330038
电 话:(0791)8678 9099
传 真:



















































©康达律师事务所 | 1988-2022 | 京ICP备09042190号-1
[北京总部] 地址:北京市朝阳区建外大街丁12号英皇集团中心8层 100022 | 电话:010-5086 7666 | 传真:010-5691 6450 | 邮箱:kangda@kangdalawyers.com
康达律师事务所(以下简称“本所”)是一家设立于中国的综合性律师事务所。本所网站上的信息仅供您参考,不应视为本所为本网站访问者就特定事项提供的法律意见或建议,本网站访问者不应将其作为作为或不作为的依据。
本所对本网站及网站所包含的文字及图片等各类信息拥有知识产权,未经授权,请勿转载或使用。
本网站超链接的第三方网站不受本所控制,仅为您方便之需,本所不对该等网站的访问者承担任何明示、默示的担保或责任。
欢迎访问本网站,如有任何问题,请与本所联系。
